Quality Control in NGS Data Analysis: A Critical First Step
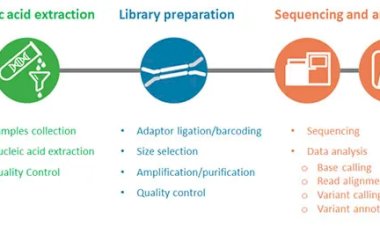
Next-Generation Sequencing (NGS) has transformed modern genomics by enabling large-scale DNA and RNA sequencing with high accuracy and speed. This blog provides a comprehensive overview of the NGS data analysis workflow, with a strong focus on quality control (QC) as a foundational step. It explains key QC parameters such as per-base sequence quality, GC content distribution, adapter contamination, and duplicate reads, along with tools used for assessment and preprocessing. The blog also discusses data trimming, reporting tools, and best practices to ensure reliable downstream analysis.
![]() Ankita Shastri
Feb 20, 2026 11:15
782
0
Ankita Shastri
Feb 20, 2026 11:15
782
0

Quality control (QC) is one of the most crucial stages in any Next-Generation Sequencing (NGS) workflow. While modern sequencing platforms generate massive amounts of data quickly and cost-effectively, raw sequencing output is rarely perfect. Technical artifacts, sequencing chemistry limitations, library preparation biases, and contamination can all introduce errors into the dataset. If these issues are not detected and corrected early, they can propagate through downstream analyses, leading to inaccurate alignments, false variant calls, misleading gene expression estimates, and ultimately flawed biological conclusions.
For this reason, QC is not just a routine checkpoint — it is a foundational process that ensures the reliability, reproducibility, and integrity of genomic research. Investing time in thorough quality assessment at the beginning saves significant effort and prevents costly mistakes later in the analysis pipeline.
Assessing Per-Base Sequence Quality
One of the first steps in QC is evaluating per-base sequence quality. During sequencing, each nucleotide is assigned a Phred quality score, which reflects the probability that a base has been incorrectly called. Higher Phred scores correspond to lower error probabilities and higher confidence in the accuracy of the read.
In many sequencing runs, especially with short-read technologies, it is common to observe a decline in quality toward the 3′ end of reads. This degradation can result from sequencing chemistry limitations or signal decay over cycles. If low-quality bases are retained, they may reduce alignment efficiency or introduce false-positive variant calls.
Per-base quality plots allow researchers to visualize the distribution of quality scores across all sequencing cycles. When quality scores drop below acceptable thresholds, trimming low-quality bases from read ends is recommended. By removing unreliable regions, researchers can significantly improve mapping accuracy and downstream analytical confidence.
Evaluating GC Content Distribution
GC content analysis is another critical component of QC. Each organism has a characteristic GC composition across its genome. Ideally, the GC distribution of sequencing reads should closely match the expected distribution for the target organism.
Significant deviations from the expected GC content may indicate contamination from another organism, biased amplification during PCR, or uneven sequencing efficiency across GC-rich or GC-poor regions. For example, extreme GC content can reduce amplification efficiency, resulting in underrepresentation of certain genomic regions.
By examining GC content distribution plots, researchers can detect abnormalities early. Consistent GC patterns across samples suggest reliable library preparation and sequencing performance, whereas unusual peaks or broad shifts may warrant further investigation.
Detecting Adapter Contamination
Adapter contamination is a frequent issue in NGS data. During library preparation, short adapter sequences are ligated to DNA or RNA fragments to enable attachment to the sequencing flow cell and amplification. If DNA inserts are shorter than the read length, sequencing may extend into the adapter region, causing adapter sequences to appear in the raw reads.
Untrimmed adapter sequences can severely interfere with downstream processes such as read alignment, de novo assembly, and variant detection. They may cause misalignments or artificially inflate error rates. Therefore, identifying and removing adapter contamination is essential for maintaining analytical accuracy.
QC tools can detect overrepresented sequences or known adapter motifs within the dataset. Once identified, trimming software can efficiently remove these unwanted sequences before further processing.
Identifying Duplicate Reads
Duplicate reads are another important QC metric. These duplicates often arise during PCR amplification steps in library preparation. While some duplication is expected, excessive duplication can indicate low library complexity or over-amplification.
High duplication rates can bias quantitative analyses. In DNA sequencing, duplicates may artificially inflate variant allele frequencies. In RNA-Seq experiments, they can distort gene expression estimates. Therefore, evaluating duplication levels helps determine whether duplicates should be marked or removed prior to downstream analysis.
Monitoring duplication rates also provides insight into overall library quality. A well-prepared library with sufficient starting material typically shows moderate duplication levels, whereas highly duplicated libraries may require re-sequencing or improved library preparation protocols.
Tools for Quality Control and Reporting
Several computational tools are widely used to perform and summarize QC analyses. FastQC is one of the most popular tools for assessing individual sequencing samples. It generates comprehensive graphical reports that include per-base quality scores, GC content distribution, sequence duplication levels, adapter content, and overrepresented sequences.
For projects involving multiple samples, managing individual QC reports can become cumbersome. In such cases, many laboratories rely on MultiQC, which aggregates outputs from multiple QC tools into a single, interactive summary report. This consolidated overview makes it easier to compare samples, detect batch effects, and identify systematic issues across sequencing runs.
Using standardized QC tools ensures consistency and reproducibility in NGS analysis pipelines.
Data Cleaning Through Trimming and Filtering
When QC analysis reveals issues such as low-quality bases or adapter contamination, preprocessing steps are applied to clean the dataset. Trimming tools such as Trimmomatic and Cutadapt are commonly used to remove adapters, trim low-quality bases from read ends, and filter out reads that fall below minimum length or quality thresholds.
Careful trimming improves read alignment rates and reduces false-positive findings. However, over-trimming can also remove useful information, so parameters must be selected thoughtfully. The goal is to strike a balance between removing unreliable data and preserving as much high-quality sequence information as possible.
After trimming, it is good practice to rerun QC to confirm that the issues have been resolved. This iterative approach ensures that the final dataset is robust and ready for downstream analysis.
The Importance of Quality Control in Reproducible Research
Beyond technical accuracy, QC plays a vital role in reproducibility. Transparent reporting of QC metrics, trimming parameters, and preprocessing decisions allows other researchers to replicate and validate findings. In clinical and large-scale genomic studies, maintaining rigorous QC standards is especially critical, as results may directly influence medical or biological interpretations.
Comprehensive documentation of QC results also facilitates troubleshooting and long-term data management. As sequencing projects grow in scale, standardized QC practices become essential for maintaining data integrity across multiple studies and institutions.
IBRI Noida’s NGS Data Analysis Program
If you’re looking to gain structured, practical training in NGS data analysis, the Indian Biological Sciences and Research Institute (IBRI) in Noida offers specialized programs tailored to students and professionals interested in genomics and bioinformatics. IBRI’s NGS training is designed to bridge the gap between theoretical knowledge and hands-on computational expertise by walking participants through real-world sequencing data workflows.
The training focuses on industry-standard tools and techniques used in modern NGS analysis workflows — including quality control, read preprocessing, sequence alignment, variant calling, and functional annotation — equipping learners with the skills needed to analyze DNA and RNA sequencing datasets effectively. IBRI emphasizes practical, dry-lab experience using real datasets, which helps participants build confidence with commonly used bioinformatics software and streamline their proficiency in handling genomic data.
In addition to hands-on NGS analysis, the institute offers a broader suite of bioinformatics and computational biology training that supports career development in fields like genomics research, personalized medicine, and computational biology. Learners at IBRI can also access guidance on related topics such as structural and functional genomics, proteomics analysis, and phylogenetics, making it a comprehensive resource for anyone looking to deepen their expertise in modern life-science data analysis.
Whether you are a life-science student, a research professional, or someone transitioning into bioinformatics, IBRI’s NGS program provides a solid foundation in both the theoretical and practical aspects of sequencing data analysis, helping you apply these workflows confidently in research or industry settings.
Conclusion
Quality control is far more than a preliminary formality in NGS data analysis — it is the backbone of trustworthy genomic research. By systematically assessing per-base sequence quality, GC content distribution, adapter contamination, and duplication levels, researchers can identify and correct technical issues before they impact downstream analyses.
With the help of tools such as FastQC and MultiQC, along with effective trimming and filtering strategies, scientists can transform raw sequencing reads into high-quality datasets suitable for accurate alignment, variant detection, and gene expression analysis.
In the rapidly evolving field of genomics, robust quality control ensures that biological insights are built on a strong and reliable foundation.